
Dear Friends,
It has been a long seven months since my last post and for that I must apologize. You see life has a funny way of getting very complicated very fast. Let me give you a summary of the events that occurred in my world over the past few months.
1. Near the end of October, my father passed away after a two and a half year battle with lung cancer (non-smoker, living only 58 years). I’ve been spending a lot of time with my mom trying to help her out as best I can.
2. Our group (myself included) have published several articles regarding the construction of trifluoromethylketones (TFMKs). Below are schemes with the associated references:
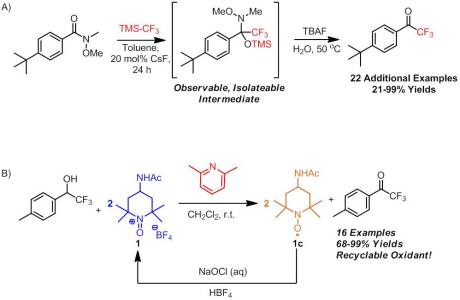
a) A Weinreb amide approach to the synthesis of trifluoromethylketones Rudzinski, D. M.; Kelly C. B.;
Leadbeater, N. E. Chem. Commun. 2012, 48, 9610
B) Oxidation of α-Trifluoromethyl Alcohols Using a Recyclable Oxoammonium Salt Kelly, C. B;
Mercadante, M. A.; Hamlin, T. A.; Fletcher, M. H.; Leadbeater, N. E. J. Org. Chem., 2012, 77, 8131
3. We are in the process of wrapping up our work with Professor Tilley as well as some other small projects to be published in the near future 🙂
4. I successfully submitted and defended my general exam. I am now a Ph.D. candidate!
5. Dr. G. K. S. Prakash visited UConn as part of the PLU-sponsored seminar series. He gave an excellent talk and I was so happy to meet one of the leaders in organofluorine chemistry!! A lot of planning went into his visit.
6. In the little time that I have had I’ve published several chemspider articles…and only a mere week ago I received an email informing me that I had won a chemspider lab coat for my posts LINK
7. We are currently working on a collaboration with Vapourtec for a educational series flow unit.
8. Our group attended the ACS national meeting in Philadelphia in August. There we meet up with one of our former undergraduate student and had an amazing time!
9. In a need to find some relaxation, I’ve been playing quite a bit of airsoft and making videos of the games. Here’s a link to one: LINK
10. I’ve applied for a summer internship at Boehringer Ingelheim to work as a research assistant at their Ridgefield, CT plant. There is a good chance I will get to work there this summer! 🙂
So as you can see, life essentially wanted all of my time. Anytime I tried to sit down and write a post I was interrupted and, rather than putting out poor posts, I decided to let the waters calm a bit. This week, I have taken time off to be with my mom on this first Christmas without my father. It has been rough for us but we’ve managed to make it a decent Christmas. Now that my house is quiet and I have far less to do than over the past seven months, I finally can sit down and just read the literature and return to posting. Rather than doing a new article, I figured I could go through our two articles that we published and then in my next post begin again fresh. So…what did we do?
In the first of the articles is the work I did with DiAndra on a one-pot route to TFMKs. The idea for this project came to me one day after reading a review on TMS-CF3. The review stated that to date, TMS-CF3 had not been used constructive manner for an acyl substitution reaction with amides. I was kind of surprised to find this because of the several successful reports of ester displacement. However, at the same time, it kind of made sense. Even if the nitrogen species was displaced, it would likely add right back into the electrophilic CF3 ketone. Thinking to basic organic chemistry, I pondered whether Weinreb amides (which have been immensely useful in constructing ketones via with Grignard reagents) would be suitable candidates as leaving group. In more general terms, I wanted to see if any amide, even a reactive one, could be used to construct TFMKs directly.
So DiAndra and myself began exploring the feasibility of this reaction. We hit immediate success with the simplest system, a phenyl Weinreb amide. By GCMS analysis we found that we had complete conversion to the TFMK. However, conversions are always deceptive and apparently the heat of the injector decomposed an unbeknownst to us intermediate When we attempted isolation by our traditional method we obtained very little TFMK. Thinking that our low yield was due to the volatility of this compound, we switched to a p-t-butyl phenyl system. Again, after workup we obtained low yield. Examining the crude reaction mixture by NMR revealed the presence of a silylated intermediate. However, as we monitored the stability of this species in THF, we found that it would slowly revert back to the starting material. Fortunately if cleaved rapidly, we could obtain substantially more CF3 ketone product. Still, yield was lacking. I then suggested that rather than go for the TFMK, let’s optimize the reaction for the silylated intermediate. Sure enough we found that, using toluene as the solvent and CsF as the fluoride source to initiate the trifluoromethylation process, we could obtain near quantitative conversion and excellent yield of the silylated intermediate. Yes, we could in fact isolate the intermediate!!

We then focused on cleavage conditions. This took a bit a work and I’ll spare you all the details. We found that heating the reaction mixture to 50 oC in the presence of TBAF in THF and H2O we could successfully convert the silylated intermediate to the TFMK product in good yield (81% for the p-t-butyl). With this two-step, one pot protocol hammered out, we moved on to a substrate screen. We found that this reaction had a reasonably broad scope, with some exceptions. Ortho-substituted arenes and straight chain systems with significant alpha branching failed to convert to the desired intermediate. Beta branching was less of a problem but did lead to lower yields. We attempted this to a steric requirement by the reaction rather than an electronic rational. My current theory is that there is a necessary chelation of TMS-CF3 by the amide Weinreb amide which is decomposed by the addition of fluoride. In some substrates, either the chelation is too poor or the activation energy for complex formation is too high and hence trifluoromethylation cannot occur.

We also explored α,β-unsaturated Weinreb amides. Here another complication arose. While we could successfully convert to the silylated intermediate quite easily, we obtained low isolated yield of the TFMK product. This time we analyzed the crude cleavage mixture and found that in addition to the desired α,β-unsaturated TFMK we found another TFMK ketone product. This new product was the result of the Michael addition of the displaced N,O-dimethylhydroxylamine anion into the highly electrophilic alkene of the desired α,β-unsaturated TFMK. Luckily, this unwanted byproduct could be removed by column chromatography. And that’s about it. What I loved most about this reaction is the scalability. One of my biggest pet peeves is when papers publish reactions done on 0.1 mmol scale. To me that’s not practical other than to maybe a chemist performing a total synthesis. This reaction has been performed anywhere from 10 mmol to 60 mmol and could likely be performed on the hundreds of mmol scale. Additionally, we have recently made some modifications that have improved yields and reaction times which will be published in chemspider in the coming week or so.
Moving on to the next article, we have a more traditional approach to TFMK construction via the oxidation of their corresponding carbinols. Not surprisingly, we attempting to do so via our favorite oxidizing agent in the Leadbeater lab, Bobbitt’s salt. To date no reports of oxidation of these difficult-to-oxidize carbinols via an oxoammonium salt have been reported. TEMPO-based methods have been reported but these protocols use aqueous media in their reaction conditions. While this is not normally a problem for traditional ketones, trifluoromethylketones have the nasty habit of hydrating in aqueous systems, particularly in basic conditions (and TEMPO oxidations are conducted under alkaline conditions). The only reliable method for oxidizing these carbinols that is currently known is the use of Dess-Martin periodinane. While I LOVE DMP, it is insanely expensive and making it in-house is…difficult…to say the least. Therefore, we set out to attempt to use Bobbitt’s salt to oxidize these alcohols. However, using the traditional conditions (DCM, SiO2, Oxidant) no reaction occurred. The putative hydride transfer from the α-carbon was likely too high in energy because of the resulting destabilized “electron-deficient” carbocation.

After some discussion with Dr. Bobbitt, we attempted to use the newer, basic conditions for our oxidation. According to Bailey and Bobbitt, the presence of a base dramatically alters the reaction mechanism. The change in reactivity results from a tightly bound ion pairing between the now formally deprotonated alcohol and the salt. This enables a more facile hydride transfer. Note that by “formally deprotonated”, I mean that there is a much greater percentage of the alkoxide in solution than under neutral/slightly acidic conditions. The bases we use, pyridyl bases, can most certainly not deprotonate the alkoxide irreversibly. We were pleased to find that under basic conditions, we could obtain quantitative conversion to the desired ketone and isolated yield. We found that as we increased the basicity of the base, the rate of the reaction increased (e.g. 2,4,6 collidine reacted faster than 2,6 lutidine) However, for in an effort to balance ease of purification with timely oxidation, we chose 2,6 lutidine over pyridine and 2,4,6 collidine.
Once we had optimal conditions, we move on to oxidizing a variety of carbinols. Aryl substituted, alkenyl, and propargyl CF3 carbinols all oxidized easily and in good yield. However, under our original conditions aliphatic CF3 alcohols failed to oxidize. To circumvent this problem, we chose to use 1,5-diazabicyclo(4.3.0)non-5-ene (DBN) as the base to formally deprotonate these alcohols. This resulted in a rapid, mildly exothermic reaction and smoothly converted the aliphatic species to TFMKs. Due to degradation of the base by the oxidant, not only was more salt required for complete conversion but more extensive purification was needed (e.g. vacuum distillation). Therefore, while it can be done with these compounds, this method likely isn’t the optimal way to oxidize CF3 alcohols lacking a neighboring sp or sp2 center.
To further demonstrate the power of our method, we found that we could selectively oxidze alcohols by simply adjusting the conditions. We conducted the following:
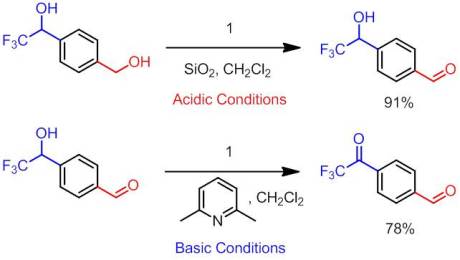
By taking advantage of the fact that the oxidant will not react with CF3 carbinols unless under basic conditions, we can selectively oxidize the non-CF3 alcohol. We can then oxidize the CF3 alcohol using our optimal conditions. Now I know what you are going to ask, what happens if you oxidize the diol under basic conditions? Unfortunately there is no selectivity; you get a mix of oxidation products.
Finally we conducted a simple rate study to get a idea of the relative rate of oxidation. We used the convenient method of Mullet and Nodding to obtain kinetic data. From there we used one of our aryl systems as our base line for the rate of oxidation. We found that the more extensive the conjugation, the fast the rate. Interestingly this was not the only factor. The propargyl CF3 system we studied oxidized extraordinarily fast (120 times faster!) indicating to us that a steric component to the oxidation was present as well. Therefore, we suggested that, in addition to cation stability, the combined electronic repulsion by the highly electron-rich CF3 group and the steric repulsion by any β-substituent contribute significantly to oxidant/alcohol complexation and hence successful oxidation. Again what I like most about our method is it’s scalability. You can perform these oxidations on any size scale. Moreover they are colorimetric. Prior to addition of the pyridyl base, the reaction will be bright yellow from the oxoammonium salt. After the base is added the reaction will transition from yellow to orange to finally deep blood red. This red color typically indicates reaction completion. Finally, you can recover the spent oxidation and reuse it to make more salt!
Well that’s it for this post. I really hoped you enjoyed the update and the work we have done over the past few months. I have to say I am very proud of what our group has accomplished so far and more is yet to come! I plan on posting much more frequently now so keep a look out for an upcoming post! Ckellz…Signing off…




