Halocycloalkenones as Diels−Alder Dienophiles. Applications to Generating Useful Structural PatternsRoss, A. G.; Townsend, S. D.; Danishefsky, S. J. J. Org. Chem. ASAP Nov. 14 2012
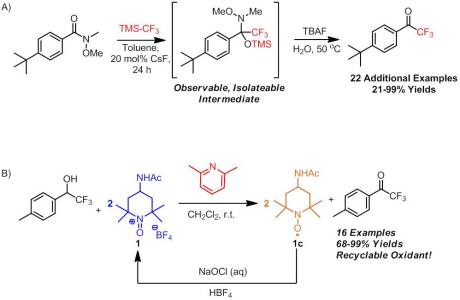
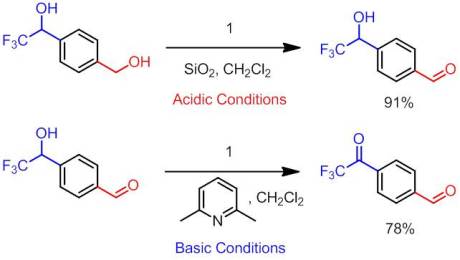
As promised, I have a review for today, the last one of 2012! I was all set to go trap shooting with my brand new shotgun with my best friend today but unfortunately mother nature had other plans. Since I’m all snowed in here, I figured there was no better time to do a review! Not much has changed in the few days since my last post with the exception of some new ChemSpider posts (Oxidation of CF3 Alcohols by an Oxoammonium Salt, Oxidation of a Propargyl Alcohol with an Oxoammonium Salt, and Synthesis of a CF3 Ketone via Trifluoromethylation of a Weinreb Amide). Go check them out! They are all based on our recently published work so I hope you enjoy them. After spending a good hour or so looking through my favorite journals and catching up on many excellent articles I missed, I decided to go with an easy pick by a legendary chemist, Samuel Danishefsky. While I’ve never had the formal pleasure of meeting Professor Danishefsky, I have seen his laboratory while I was down at Columbia. He certainly has a nice set-up down there. Moreover, as the director for the Bioorganic Chemistry Laboratory at Sloan-Kettering Cancer Research Institute, much of his work is directed towards targeted syntheses . In fact, my former graduate student adviser at Columbia (who came to my talk at the ACS conference in Philadelphia!) currently works as a NIH post-doc at Sloan under Professor Danishefsky. While much of Danishefsky’s work centers around total synthesis, he is no foreigner to methods development. I’ve said it before but I will say it again: the best way to figure out where methods are sorely needed is by conducting a targeted synthesis. It’s quite clear that Danishefsky knows this concept quite well. Danishefsky is most famous for his named diene which has seen widespread usage because of its highly regioselective additions in Diels-Alder reaction.
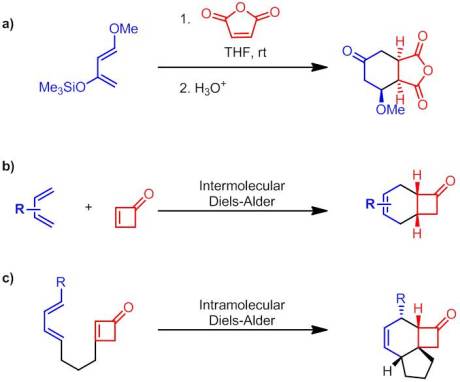
a) Typical Danishefsky’s Diene Diels-Alder Reaction b) Intermolecular Cyclobutenone Diels-Alder Reaction c) Intramolecular Cyclobutenone Diels-Alder Reaction
On a related note, Danishefsky has recently been exploring the Diels-Alder of chemistry of an interesting dienophile, cycloalkenones. Specifically, he has been focusing on cyclobutenones as coupling partners both in the inter– and intra-molecular sense. This new area of research the Danishefsky group is pursuing is part of a more general strategy to promote pattern recognition analysis (PRA). PRA provides an interesting alternative to Corey’s strategic bond-oriented retrosynthetic approach to total synthesis. PRA takes more of a building block approach and hence the key to PRA is having a diverse array of “templates”, or core motifs as he calls them, to build off of.
Danishefsky’s explorations in cyclobutenone chemistry have proven quite successful in diversifying the available templates. However, there is always room for improvement in any type of chemistry. Seeking to “enhance the synthetic value” of cyclobutenones, his attention turned to 2-halocycloalkenones. Having developed a reliable route to synthesize cyclobutenone, preparation of its 2-bromo derivative was quite easy. Treatment with molecular bromine followed by an E2 elimination affords the vinylic bromide in 67% overall yield.
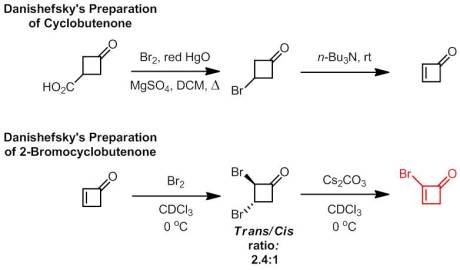
As a side note, I was quite curious about cyclobutenone. Surprisingly, or maybe not surprisingly, it’s not all that stable and readily polymerize at room temperature. It can be stored neat at -78 oC for a very short period of time but apparently can be kept as a stock solution in deuterated chloroform for much longer. I was also surprised that it was not until 1971 that it was definitively synthesized by Sieja and, even after it was synthesized, the chemical community paid little attention to it. To me, being a classical physical organic chemist, I thought this was somewhat of a travesty considering the interesting properties such a strained molecule must have!
After preparing his model 2-halocyclobutenone system, Danishefsky then began his investigation of it ability to serve as a dienophile. He was delighted to find that using a variety of dienes he could obtain excellent yields of the corresponding bicyclic (or in one case tricyclic) adducts. Moreover, the reaction was diastereospecific, producing only a single pair of diastereomers. He was even able to produce a facile reaction with Dane’s diene.
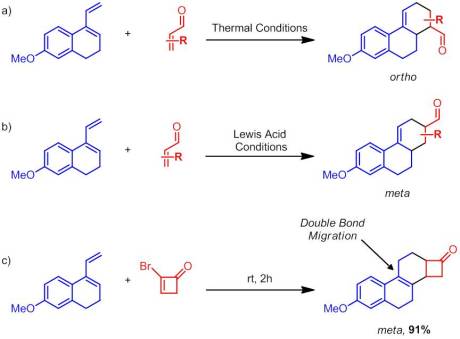
a) Dane’s diene reacting with a dienophile under thermal conditions b) Dane’s diene reacting with a dienophile under Lewis acidic conditions c) Dane’s diene reacting with a 2-bromocyclobutenone
I’ll confess that I was unfamiliar with Dane’s diene until reading this paper. According to Danishefsky, Dane’s diene doesn’t undergo Diels-Alder chemistry all too easily. When it does it gives two distinct products depending on the conditions. If Lewis acid catalysis is used the “meta” product is obtained while the “ortho” product is produced under thermal conditions:
Using 2-bromocyclobutenone, strictly the “meta” product was obtained in excellent yield. Additionally, the reaction was complete in only two hours at room temperature! During the course of the reaction, the double bond undergoes an unusual migration to become tetrasubstituted. No explanation for how this migration occurred is given, though my guess is that the isomerization reaction is low enough in energy (and produces the more thermodynamically stable alkene) that it can occurs spontaneously.

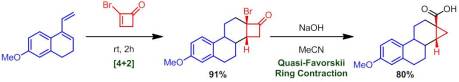
Danishefsky then moved on to assess the reactivity of larger 2-halocycloalkenones. He found that unlike their non-halogenated analogues, 2-bromocyclopentenone and 2-bromocyclohexenone reacted quite readily and gave good to excellent yield of their corresponding Diels-Alder adducts. Treatment of these adducts with sodium hydroxide in MeCN gave, at least to me, the most synthetically interesting products. These adducts underwent a quasi-Favorskii ring contraction (one of my favorite named reactions) to yield cyclopropane-containing products:
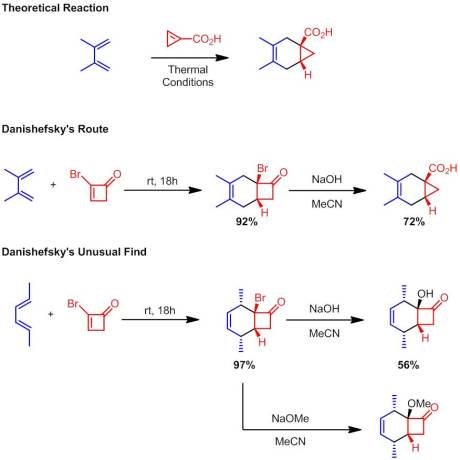
Danishefsky totes this as an alternative way to access the theoretical product of the Diels-Alder reaction between a cyclopropenyl carboxylic acid and a diene. Interestingly, one of the compounds examined does not undergo a ring contraction but rather gives the α-hydroxy substitution product … with retention of stereochemistry! Even more unusually is that treatment with methoxide produces an analogous result, the α-methoxy substitution product. Now I’m going to do something I normally don’t do here at New Reactions: put my own two cents in about an unusual finding in an article. I’m going suggest a mechanism that’s consistent with the observed data but…WARNING: I have no proof for this mechanism and some of it may be heretical. My hope is that we can maybe get a bit of discussion going out of it:
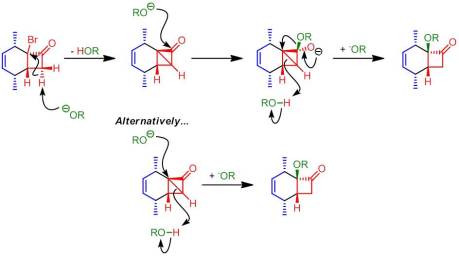
So what do you think? What are your thoughts on this peculiar product?
Well I hope you enjoyed this review and I recommend you take a look at the article yourself. Hats off to Danishefsky and co-workers for an excellent, well-written, and thorough article. I look forward to more work on these 2-halocycloalkenones! Ckellz…Signing off…